pkgs <- c("fs", "futile.logger", "configr", "stringr", "ggpubr", "ggthemes", "SingleCellExperiment",
"RColorBrewer", "vroom", "jhtools", "glue", "jhuanglabHyperion", "openxlsx", "ggsci", "ggraph",
"patchwork", "cowplot", "tidyverse", "dplyr", "rstatix", "magrittr", "igraph","tidygraph")
suppressMessages(conflicted::conflict_scout())
for (pkg in pkgs){
suppressPackageStartupMessages(library(pkg, character.only = T))
}
res_dir <- "./results/sup_figure7" %>% checkdir
dat_dir <- "./data" %>% checkdir
config_dir <- "./config" %>% checkdir
#colors config
config_fn <- glue::glue("{config_dir}/configs.yaml")
stype3_cols <- jhtools::show_me_the_colors(config_fn, "stype3")
ctype10_cols <- jhtools::show_me_the_colors(config_fn, "cell_type_new")
meta_cols <- jhtools::show_me_the_colors(config_fn, "meta_color")
#read in coldata
coldat <- readr::read_csv(glue::glue("{dat_dir}/sce_coldata.csv"))
meta_clu <- readxl::read_excel(glue::glue("{dat_dir}/meta_clu.xlsx")) %>% dplyr::select(-9)
ci <- readr::read_rds(glue::glue("{dat_dir}/ci_list.rds"))
sample_chemo_type_list <- readr::read_rds(glue::glue("{dat_dir}/sample_chemo_type_list.rds"))
metadata <- readr::read_rds(glue::glue("{dat_dir}/metadata.rds"))
# function
interaction_plot <- function(x, conditionCol, group, colors, threshold = 0.1, selfDe = T){
from <- "from_meta"
to <- "to_meta"
fn <- "metacluster_"
cols <- colors
if (selfDe == TRUE) {
fil <- "filteredself"
dt_cl <- x %>% dplyr::filter(.data[[from]] != .data[[to]]) %>%
group_by(.data[[from]], .data[[to]], .data[[conditionCol]]) %>%
summarise(N = n()) %>% left_join(x %>% dplyr::filter(.data[[from]] != .data[[to]]) %>%
group_by(.data[[conditionCol]]) %>%
summarise(NT = n()), by = conditionCol) %>%
dplyr::mutate(pct = N/NT * 100)
} else {
fil <- ""
dt_cl <- x %>% group_by(.data[[from]], .data[[to]], .data[[conditionCol]]) %>%
summarise(N = n()) %>% left_join(x %>% group_by(.data[[conditionCol]]) %>%
summarise(NT = n()), by = conditionCol) %>%
dplyr::mutate(pct = N/NT * 100)
}
dt_cl <- dt_cl %>% group_by(.data[[conditionCol]])
glist <- dt_cl %>% dplyr::filter(pct > 0.1) %>%
group_map(~as_tbl_graph(dplyr::select(.,c(.data[[from]], .data[[to]], pct))))
gkeys <- dt_cl %>% group_keys()
plist <- list()
for(i in 1:length(glist)){
g <- glist[[i]]
if(length(g) == 0) next
names <- paste0(fn, group, "_", gkeys[i,][[conditionCol]], "_", fil)
plist[[names]] <- ggraph(g, "stress") +
geom_edge_link(aes(edge_width = pct, alpha = pct)) +
geom_node_point(aes(color = name), size = 3) +
#geom_node_text(aes(label = name), size = 4, repel = T, colour = "red") +
scale_edge_width("interaction percentage", range = c(0.2, 1.5),
breaks = c(0.1,1,3,5,10,15,20,25),limits=c(0.1, 25), #oob = scales::squish,
guide = guide_legend(title.theme = element_text(size = 8),
ncol = 1, byrow = FALSE, keywidth = 0.8,
keyheight = 0.1)) +
scale_edge_alpha("interaction percentage", range = c(0.1, 1.5),
breaks = c(0.1,1,3,5,10,15,20,25),
limits=c(0.1, 25), oob = scales::squish) +
scale_color_manual(limits = as.factor(V(g)$name), values = colors) +
theme_graph(base_family = "Helvetica", base_size =8) +
guides(col = guide_legend(title = "interaction", ncol = 1,
byrow = FALSE, keywidth = 0.1, keyheight = 0.1,
override.aes = list(size=1),
title.theme = element_text(size = 8)))
}
return(plist)
}sup_figure7
sup_figure7a
keycols_list <- c(list("nerve_invasion"), list("vascular_invasion"),
list("lymph_metastasis_status"), list("size_alteration"),
list("size_alteration"), list("pfs_group"))
compare_groups <-c("nerve_invasion", "vascular_invasion", "lymph_metastasis",
"chemo_outcome_before", "chemo_outcome_after", "pfs_analysis")
names(keycols_list) <- compare_groups
meta_cluster_names <- rev(c("MC-macro-c2", "MC-macro-c1", "MC-immune-myeloid",
"MC-immune-enriched", "MC-stroma-CAF", "MC-stroma-macro",
"MC-stroma-mCAF", "MC-tumor-frontline", "MC-tumor-core"))
total_group <- coldat %>%
dplyr::select(c(sample_id, sample_tiff_id, patient_id, cell_id, stype2,
cluster_names, meta_cluster)) %>%
na.omit() %>% dplyr::filter(meta_cluster %in% meta_cluster_names)
factor_list <- list("nerve_invasion" = c("1", "0"),
"vascular_invasion" = c("1", "0"),
"lymph_metastasis" = c("1", "0"),
"chemo_outcome_before" = c("increase", "decrease"),
"chemo_outcome_after" = c("increase", "decrease"),
"pfs_analysis" = c("short","long"))
plist <- list()
for(compare_group in compare_groups){
#cli::cli_h1(compare_group)
groups <- metadata[[compare_group]]
gp_key <- keycols_list[[compare_group]]
cli::cli_alert_info(gp_key)
groups <- groups %>% drop_na(all_of(gp_key))
dt_groups <- total_group %>% dplyr::filter(patient_id %in% groups$patient_id)
if (compare_group == "chemo_outcome_before") {
dt_groups <- dt_groups %>% dplyr::filter(stype2 %in% c("before_chemo","puncture_pdac"))
} else if (compare_group %notin% c("paired_after_neoadj", "mpc_primary_metastasis", "tumor_para")) {
dt_groups <- dt_groups %>% dplyr::filter(stype2 %in% c("after_chemo","tumor"))
}
dat_plot <- dt_groups %>%
group_by(meta_cluster, sample_tiff_id) %>%
summarise(nc = n()) %>%
group_by(sample_tiff_id) %>%
dplyr::mutate(nt = sum(nc)) %>%
dplyr::mutate(pro = nc/nt) %>% ungroup()
dat_plot$meta_cluster <- factor(dat_plot$meta_cluster, levels = meta_cluster_names)
metainfo <- coldat[, c("sample_tiff_id", "patient_id")] %>% distinct()
dat_plot <- inner_join(dat_plot, metainfo, by = "sample_tiff_id") %>%
inner_join(groups, by = "patient_id") %>% distinct()
dat_plot[[gp_key]] <- factor(dat_plot[[gp_key]], levels = factor_list[[compare_group]])
dat_plot$meta_cluster <- fct_relevel(dat_plot$meta_cluster, meta_cluster_names)
p <- ggboxplot(dat_plot, x = "meta_cluster", y = "pro", fill = gp_key, outlier.shape = NA,
palette = pal_nejm("default")(3), xlab = NULL,size = 0.2) + theme_classic() +
theme(strip.placement = "outside",
panel.spacing = unit(3, "points"),
strip.background = element_blank(),
strip.text = element_text(face = "bold", size = 5),
axis.text.x = element_text(size = 10), axis.text.y = element_text(size = 10),
axis.line.x = element_line(size = 0.4),
axis.line.y = element_line(size = 0.4),
legend.position="right") +
labs(x= NULL, y = NULL)
exp1 <- expr(pro ~ !!ensym(gp_key))
stat_test <- dat_plot %>%
group_by(meta_cluster) %>% rstatix::wilcox_test(eval(exp1), p.adjust.method = "none")
stat_test <- stat_test %>% mutate(p.adj.signif = case_when(p >= 0.05 ~ "ns",
p >= 0.01 & p < 0.05 ~ "*",
p >= 0.001 & p < 0.01 ~ "**",
p >= 0.0001 & p < 0.001 ~ "***",
p < 0.0001 ~ "****",
TRUE ~ "ns"))
stat_test <- stat_test %>%
add_xy_position(x = "meta_cluster", dodge = 0.8)
p1 <- p +
stat_pvalue_manual(
stat_test, tip.length = 0.01, hide.ns = T, label = "p.adj.signif",
coord.flip = TRUE
) + coord_flip() +
guides(fill = guide_legend(reverse = TRUE))
#ggsave(glue("metacluster_{compare_group}.pdf"), p1, width = 6, height = 6)
plist[[compare_group]] <- p1
}
pc <- (plist[[1]]|plist[[2]]|plist[[3]])/(plist[[4]]|plist[[5]]|plist[[6]]) +
plot_layout(guides = 'collect', widths = c(2, 2.5, 2.5, 2.5))
ggsave(glue::glue("{res_dir}/sup_figure7a_wilct.pdf"), pc, width = 10.5, height = 5)
pc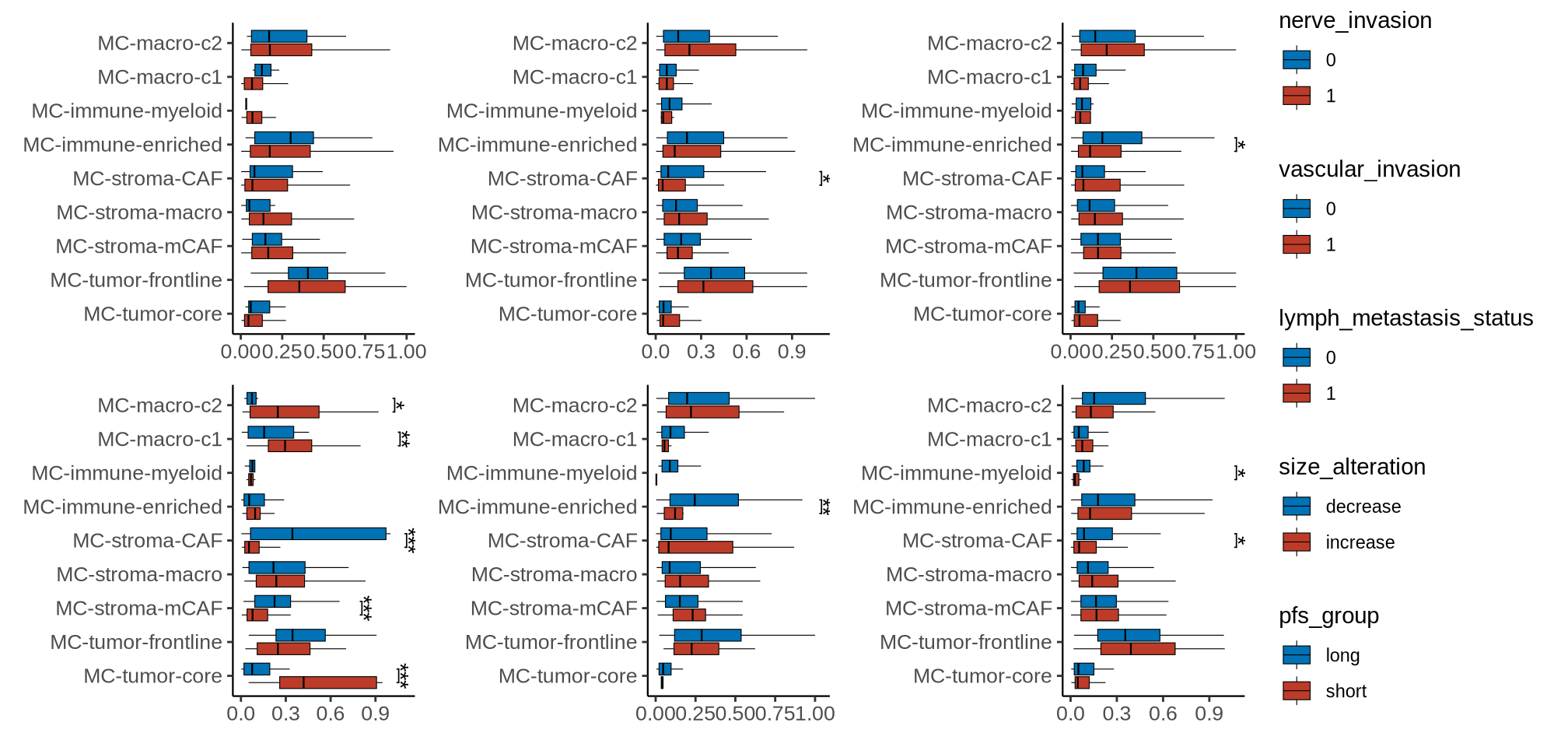
sup_figure7b
# clinical metadata
#chemo_outcome_before
dt_cob <- metadata[["chemo_outcome_before"]][c("patient_id", "size_alteration")]
dt_cob <- coldat %>% dplyr::filter(patient_id %in% dt_cob$patient_id) %>%
dplyr::select(sample_id, stype2, patient_id, sample_tiff_id) %>% unique() %>%
left_join(dt_cob, by = "patient_id") %>%
dplyr::filter(stype2 %in% c("before_chemo", "puncture_pdac"))
#chemo_outcome_after
dt_coa <- metadata[["chemo_outcome_after"]][c("patient_id", "size_alteration")]
dt_coa <- coldat %>% dplyr::filter(patient_id %in% dt_coa$patient_id) %>%
dplyr::select(sample_id, stype2, patient_id, sample_tiff_id) %>% unique() %>%
left_join(dt_coa, by = "patient_id") %>%
dplyr::filter(stype2 %in% c("after_chemo", "tumor"))
dt_list <- list(dt_cob, dt_coa)
names(dt_list) <- c("chemo_outcome_before", "chemo_outcome_after")
for (i in 1:length(dt_list)) {
dt_list[[i]] <- left_join(dt_list[[i]], do.call("rbind", ci),
by = "sample_tiff_id") %>% dplyr::select(-c(from, to))
dt_list[[i]] <- dt_list[[i]] %>% left_join(meta_clu[, c("old_cluster_name", "meta_short_new")],
by = c("from_cluster" = "old_cluster_name")) %>%
dplyr::rename("from_meta_old" = "from_meta", "from_meta" = "meta_short_new") %>%
left_join(meta_clu[, c("old_cluster_name", "meta_short_new")],
by = c("to_cluster" = "old_cluster_name")) %>%
dplyr::rename("to_meta_old" = "to_meta", "to_meta" = "meta_short_new")
dt_list[[i]][["size_alteration"]] <- factor(dt_list[[i]][["size_alteration"]],
levels = factor_list[[names(dt_list)[i]]])
}
group_list <- list(c("chemo_outcome_before", "size_alteration"),
c("chemo_outcome_after", "size_alteration"))
p1 <- purrr::map2(dt_list, group_list, ~interaction_plot(x = .x, conditionCol = .y[2],
group = .y[1], colors = meta_cols))
names(p1) <- c("chemo_outcome_before", "chemo_outcome_after")
#sub keycols
keycols_sub <- keycols_list[c("nerve_invasion", "vascular_invasion", "lymph_metastasis", "pfs_analysis")]
dt_list <- list()
for (i in 1:length(keycols_sub)) {
dt_list[[i]] <- metadata[[names(keycols_sub[i])]][c("patient_id", keycols_sub[[i]])]
dt_list[[i]] <- coldat %>% dplyr::filter(patient_id %in% dt_list[[i]]$patient_id) %>%
dplyr::select(sample_id, stype2, patient_id, sample_tiff_id) %>% unique() %>%
left_join(dt_list[[i]], by = "patient_id") %>% dplyr::filter(stype2 %in% c("after_chemo", "tumor"))
}
for (i in 1:length(dt_list)) {
dt_list[[i]] <- left_join(dt_list[[i]], do.call("rbind", ci), by = "sample_tiff_id") %>%
dplyr::select(-c(from, to))
dt_list[[i]] <- dt_list[[i]] %>% left_join(meta_clu[, c("old_cluster_name", "meta_short_new")],
by = c("from_cluster" = "old_cluster_name")) %>%
dplyr::rename("from_meta_old" = "from_meta", "from_meta" = "meta_short_new") %>%
left_join(meta_clu[, c("old_cluster_name", "meta_short_new")],
by = c("to_cluster" = "old_cluster_name")) %>%
dplyr::rename("to_meta_old" = "to_meta", "to_meta" = "meta_short_new")
dt_list[[i]][[keycols_sub[[i]]]] <- factor(dt_list[[i]][[keycols_sub[[i]]]],
levels = factor_list[[names(keycols_sub)[i]]])
}
p_list <- list()
for (i in 1:length(dt_list)) {
for (j in 1:length(keycols_sub[[i]])) {
group1 <- str_c(names(keycols_sub)[i],keycols_sub[[i]][j], sep= "_")
p_list[[group1]] <- interaction_plot(dt_list[[i]] %>%
dplyr::select(sample_id, keycols_sub[[i]][j], sample_tiff_id,
from_cluster, to_cluster, from_meta, to_meta) %>% na.omit(),
conditionCol = keycols_sub[[i]][j], group = group1, colors = meta_cols)
}
}
p_list <- c(p_list, p1)
lo <- "
ABC
DEF
GHI
JKL
"
p2 <- p_list[["nerve_invasion_nerve_invasion"]][[1]] + p_list[["vascular_invasion_vascular_invasion"]][[1]] +
p_list[["lymph_metastasis_lymph_metastasis_status"]][[1]] + p_list[["nerve_invasion_nerve_invasion"]][[2]] +
p_list[["vascular_invasion_vascular_invasion"]][[2]] + p_list[["lymph_metastasis_lymph_metastasis_status"]][[2]] +
p_list[["chemo_outcome_before"]][[1]] + p_list[["chemo_outcome_after"]][[1]] +
p_list[["pfs_analysis_pfs_group"]][[1]] + p_list[["chemo_outcome_before"]][[2]] +
p_list[["chemo_outcome_after"]][[2]] + p_list[["pfs_analysis_pfs_group"]][[2]] + plot_layout(design = lo, guides='collect')
ggsave(glue::glue("{res_dir}/sup_figure7b.pdf"), p2, width = 12, height = 9.5)
p2